mmBiologics
Leveraging Mass Spectrometry to Efficiently Deliver Precise and Actionable Insights into Biologics (RNA / DNA, Oligonucleotides, Conjugates, and Peptides / Proteins).
Overview
mmBiologics leverages mass spectrometry to efficiently deliver precise and actionable insights into biologics. The solution provides unified XIC generation / quantification, deconvolution and sequencing*, with visualizations for deeply interrogating datasets. Multiple datasets can be batch processed via workflows that automatically perform LC peak detection, quantitation, deconvolution and sequencing*.
Overview
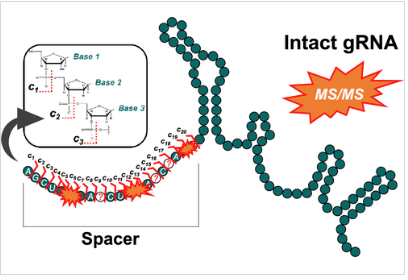

Spacer Fidelity Assessments of Guide RNA by Top-Down Mass Spectrometry
ABSTRACT: The advancement of CRISPR-based gene editing tools into biotherapeutics offers the potential for cures to genetic disorders and for new treatment paradigms for even common diseases. Arguably, the most important component of a CRISPR-based medicine is the guide RNA, which is generally large (>100-mer) synthetic RNA composed of a “tracr” and “spacer” region, the latter of which dictates the on-target editing site as well as potential undesired off-target edits. Aiming to advance contemporary capabilities for gRNA characterization to ensure the spacer region is of high fidelity, top-down mass spectrometry was herein implemented to provide direct and quantitative assessments of highly modified gRNA. In addition to sequencing the spacer region and pinpointing modifications, top-down mass spectra were utilized to quantify single-base spacer substitution impurities down to U and U > C substitutions, and created a de novo sequencing strategy to facilitate the identification and quantification of gRNA impurities with highly dissimilar spacer regions.
Explore the detailed study on Spacer Fidelity Assessments of Guide RNA by Top-Down Mass Spectrometry, featured in ACS Central Science.
NIH Grant awardee
Leveraging Mass Spectrometry to Efficiently Deliver Precise and Actionable Insights into Biologics
RNA / DNA, Oligonucleotides, Conjugates, and Peptides / Proteins
Unified XIC generation/quantification, deconvolution, and sequencing*
Full isotopic distribution determines sequence* match
Modern Cloud Delivery (yours or ours)
Sequencing
-
Complete isotopic distribution
utilized for precise sequence matching -
Fully customizable sequence definitions allow for
infinitely flexible sequences (non-natural definitions too) - Sequence analysis score comparison
- Automated Workflow
- Automatic Molecular Annotations



Deconvolution
-
Accurate deconvolution across the full
range of retention times -
Determine monoisotopic & calculated average mass
for a fully resolved intact isotopic envelope - Automated Workflow
- Automatic Molecular Annotations



XIC Generation / Quantitation
-
Generate & quantify
extracted ion chromatograms (XIC) -
Automatically determining
the dynamic charge range based on data - UV data supported
- Automated Workflow
- Automatic Molecular Annotations
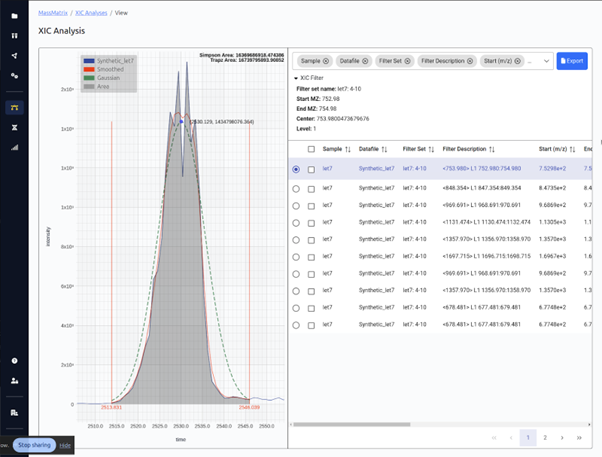

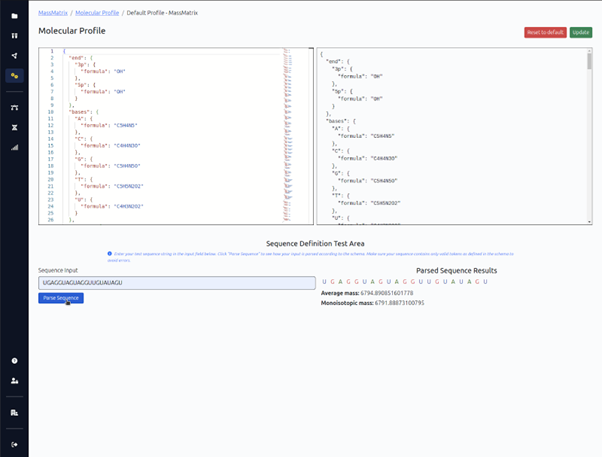
Molecular Profile
The molecular profiling page allows full customization of the molecular chemistry. Modifications can be independently added to the base, sugar, and/or linker without compromising MS/MS fragment analysis.
Custom nucleotides, sugars, and linker moieties can be modeled, so you’re not limited to naturally occurring chemistries.
- Streamlined process for generating and quantifying extracted ion chromatograms (XIC)
- Automated, fast, and accurate deconvolution across the full range of retention times
- Complete isotopic distribution is utilized for precise sequence* matching
- Flexible Cloud Delivery via your cloud infrastructure or our managed cloud service.
- Easily share data with colleagues and collaborators.
- User-friendly interface for seamless data interpretation and presentation.
- Advanced fragmentation model for oligonucleotides
- Peptide fragmentation model (in development)
- Designed to work with unique custom oligonucleotides, peptides, modifications, and other molecules.
- No hidden proprietary formats, ensuring transparency and easy access.
- An API driven architecture means all data is easily accessible outside of mmBiologics.
- Directly query the database.
- Build personalized applications or dashboards for specific data analysis needs.
mmBiologics
Unified XIC generation/quantification, deconvolution, and sequencing*
Full isotopic distribution determines sequence* match
Modern Cloud Delivery (yours or ours)
Sequence analysis provides an overview of all detections based on their ion, position, and charge state. The unique scoring algorithm scores the entire isotopic envelope, improving the detection and qualification of ion matches. The user can filter scan, charge state, score, consecutive matches, and monoisotopic matches. All plots and tables are exportable in common formats (PNG, SVG, CSV).
When viewing, the best ion match can be found by clicking on the top row. All plots and tables are exportable in common formats (PNG, SVG, CSV).
Not only are scores for each sequence analysis produced, but comparisons across charge states and scans can be made. This gives an objective comparison when studying sequences detected in various samples. All plots and tables are exportable in common formats (PNG, SVG, CSV).
XICs are presented in an intuitive format enabling quick evaluation of the XIC and setting of the RT range for calculating an accurate area under the curve. All plots and tables are exportable in common formats (PNG, SVG, CSV).
The XIC wizard allows for quickly generating an analysis of all possible XIC ranges for a given molecule.
The auto deconvolution feature provides a variety of algorithms to efficiently search MS and UV data. Fixed retention time (RT) ranges and automatic peak detection (valley to valley) of MS and UV data are available for batch signal averaging and deconvolution. Results are presented in an intuitive user interface for easy interpretation. Multiple files can be processed in parallel and distributed and scaled across cloud resources. All plots and tables are exportable in common formats (PNG, SVG, CSV).
Automatic annotations via database lookups for known molecules are provided when evaluating deconvolution results. All plots and tables are exportable in common formats (PNG, SVG, CSV).
The monoisotopic mass as well as the calculated average mass for a fully resolved intact isotopic envelope are easily viewable. All plots and tables are exportable in common formats (PNG, SVG, CSV).
The molecular profiling page allows full customization of the molecular chemistry. Modifications can be independently added to the base, sugar, and/or linker without compromising MS/MS fragment analysis. Custom nucleotides, sugars, and linker moieties can be modeled, so you're not limited to naturally occurring chemistries.
Key Advantages of mmBiologics
mmBiologics is crafted with precision and backed by extensive research. It automates data analysis workflows and enables deep data interrogation.

Best-in-Class Sequencing
Experience superior sequencing accuracy for oligonucleotides and conjugates, ensuring faster and more reliable research outcomes. Complete isotopic distribution is utilized for precise sequence matching.

Power Users & Data Scientists
- No hidden proprietary formats, ensuring transparency and easy access.
- An API driven architecture means all data is easily accessible outside of mmBiologics.
- Directly query the database.
- Build personalized applications or dashboards for specific data analysis needs.

Simplified User Interface
- Flexible Cloud Delivery via your cloud infrastructure or our managed cloud service.
- Easily share data with colleagues and collaborators.
- User-friendly interface for seamless data interpretation and presentation.
Advanced Molecule Characterization
- Advanced fragmentation model for oligonucleotides
- Peptide fragmentation model (in development)
- Designed to work with unique custom oligonucleotides, peptides, modifications, and other molecules.
Spacer Fidelity Assessments of Guide RNA by Top-Down Mass Spectrometry
ABSTRACT: The advancement of CRISPR-based gene editing tools into biotherapeutics offers the potential for cures to genetic disorders and for new treatment paradigms for even common diseases. Arguably, the most important component of a CRISPR-based medicine is the guide RNA, which is generally large (>100-mer) synthetic RNA composed of a “tracr” and “spacer” region, the latter of which dictates the on-target editing site as well as potential undesired off-target edits.
Aiming to advance contemporary capabilities for gRNA characterization to ensure the spacer region is of high fidelity, top-down mass spectrometry was herein implemented to provide direct and quantitative assessments of highly modified gRNA. In addition to sequencing the spacer region and pinpointing modifications, top-down mass spectra were utilized to quantify single-base spacer substitution impurities down to U and U > C substitutions, and created a de novo sequencing strategy to facilitate the identification and quantification of gRNA impurities with highly dissimilar spacer regions.
Explore the detailed study on Spacer Fidelity Assessments of Guide RNA by Top-Down Mass Spectrometry, featured in ACS Central Science.
Spacer Fidelity Assessments of Guide RNA by Top-Down Mass Spectrometry
ABSTRACT: The advancement of CRISPR-based gene editing tools into biotherapeutics offers the potential for cures to genetic disorders and for new treatment paradigms for even common diseases. Arguably, the most important component of a CRISPR-based medicine is the guide RNA, which is generally large (>100-mer) synthetic RNA composed of a “tracr” and “spacer” region, the latter of which dictates the on-target editing site as well as potential undesired off-target edits. Aiming to advance contemporary capabilities for gRNA characterization to ensure the spacer region is of high fidelity, top-down mass spectrometry was herein implemented to provide direct and quantitative assessments of highly modified gRNA. In addition to sequencing the spacer region and pinpointing modifications, top-down mass spectra were utilized to quantify single-base spacer substitution impurities down to U and U > C substitutions, and created a de novo sequencing strategy to facilitate the identification and quantification of gRNA impurities with highly dissimilar spacer regions.
Explore the detailed study on Spacer Fidelity Assessments of Guide RNA by Top-Down Mass Spectrometry, featured in ACS Central Science.